
3 min read
Most life sciences organizations find it difficult to streamline their Regulatory submissions process due to frequent changes in the submission guidelines. According to a recent analysis conducted by the United States Food and Drug Administration (USFDA), 32% of submissions with study data had critical data conformance issues. The process necessitates an extensive understanding of collaboration among internal and external stakeholders. Within the organization, the manufacturing, research and development, clinical/non-clinical research, QA/QC, marketing, and sales departments must work in harmony to accelerate the approval process from the initial development to when the drug hits the market. The Regulatory Affairs department and the Health Authorities (HAs) are in constant correspondence to ensure and assess the safety and efficacy of drugs for human use. Let us see what preliminary measures pharmaceutical organizations must take to establish Regulatory submissions excellence:
Allocate and deploy resources to streamline, standardize, and fastrack disorganized submission work
A submission specialist participates on a contractual basis and can comprehend more than one (01) type of submission while being associated with development programs over the years, assisting in crafting compelling narratives for the dossier. Doing so makes it easier for organizations to:
- Eliminate errors by balancing the internal and external stakeholders’ collaboration
- Reduce the HA review cycle of the document authoring and approval process
- Establish complete and compliant submissions to keep up with the changing Regulatory requirements for different markets
- Acknowledge the submissions affected by a Regulatory Authority
- Be aware of the status of submission authoring, review, and approvals
- Enhancement of the current submission protocol
Simplify dossier submissions methodology
Authoring a dossier is most crucial when it comes to the preparation of submissions. It is observed that the Biologics License Applications (BLAs) and the New Drug Applications (NDAs) comprise tons of pages, making it difficult for regulators to understand the narrative of the dossier. Having a structured representation of a general idea can amplify the team’s capability of preparing submissions and the agencies reviewing them.
Using modules to streamline submissions enables the experts to analyze what is to be included in each section or module of the dossier. Such a practice facilitates:
- Early initiation of authoring by accelerating the organization’s submission preparation
- Use of relevant data required for authoring the submissions
- Use of proper resources without the need for writing extensive documents from scratch for every submission
Work in collaboration with the Regulatory Authorities
Every organization must conduct regular meetings with the HAs as a practice during the early stage of product development. This ensures timely review and feedback, resulting in an expedited resolution of the comments by the HA, and consequently, the approval process. It not only builds a good rapport between the organizations and HA but also helps in getting clarity at an early stage of submission planning.
Streamline content and data in the Document Management System (DMS)
A robust content repository is expected to deal with all the data across functional areas, including the documentation and information created previously. Structured content and data repository provide seamless access to documents from various functional areas without any extra effort or the need to duplicate. Documents stored in the DMS become the single source of contact for all the internal stakeholders (clinical science, CMC, nonclinical, pharmacokinetics, pharmacodynamics, medical writing, and clinical operations). Such content and data can be utilized to create local content based on regional requirements. Optimizing Regulatory Submissions via Structured Content and Data Management
Adopting technology and automation to reduce repetitive work and errors
Organizations can make use of technology by using its auto-update and quality control of the in-text Tables, Listings, and Figures (TLF) against the source data feature to accelerate the documentation process. In this way, whenever there are any changes made to the existing source tables, there is an update made in the in-text TLF accordingly. This helps eliminate overall work hours, save time on the critical path, and enhance data accuracy.
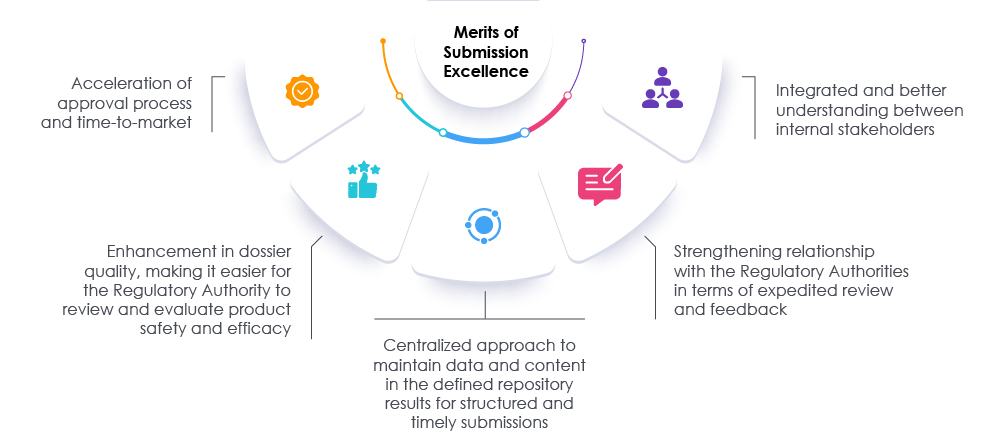
Adopting appropriate measures along with timely reviews of the dossiers and the data can help both regulators and organizations to speed up the submission process. This approach accelerates the time to market so as to meet the needs of the patients as well as the industry. A proven Regulatory partner can assist organizations in rethinking, reassessing, and modifying the current approaches for preparing submissions. Our experts at Freyr can facilitate a path towards attaining Regulatory submissions’ excellence by increasing visibility across the organization and promoting overall compliance, allowing the organization to succeed in today’s life sciences market. Consult Freyr.









