


Medical Device Registration in Indonesia Overview
Indonesia has initiated universal health care for their citizens in 2014. This has greatly influenced the growth of Medical Devices market and led to the rise in the Medical Devices import. The devices in Indonesia are regulated by the National Agency of Drug and Food Control (NADFC) functioning under the Indonesian Ministry of Health (MoH). The latest regulation in place for import of Medical Devices is Decree No. 62 imposed in the year 2017. Foreign companies must appoint an Indonesia Local authorised representative for the process of Medical Device Registration in Indonesia.
![]()
Regulatory Authority: National Agency of Drug and Food Control (NADFC)![]()
Regulation: No. 62 / 2017![]()
Authorized Representative: Indonesia Local Authorised representative![]()
QMS Requirement: ISO 13485:2016![]()
Assessment of Technical Data: NADFC![]()
Validity of License: 5 Years![]()
Labeling Requirements: No. 62 / 2017![]()
Submission Format: Online/Paper![]()
Language: English & Indonesian
Indonesia Medical Device Classification
The current regulation classifies the devices as A, B, C, and D based on risk.
| Risk Criteria | Device Class |
|---|---|
| Low Risk | A |
| Low Moderate Risk | B |
| Moderate – High Risk | C |
| High Risk | D |
Indonesia Local Authorised Representative
The Indonesian regulations require the manufacturers to appoint a local representative with a Distributor License. A distributor can be appointed to represent the foreign manufacturer in Indonesia. However, appointing an independent third party would provide flexibility to change distributors or appoint multiple distributors for better market penetration.
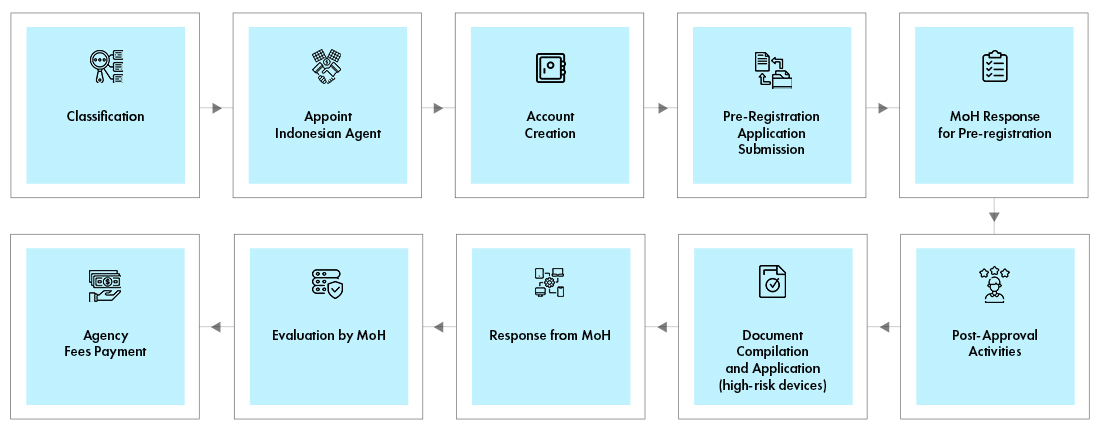
Indonesia Medical Device Registration
The Local Representative must create an account in the online portal. The registration process is same for all the device classes. However, the documentation requirement varies with class of device. The registration is a two stages process –
- Pre-Registration process
- Evaluation Process
MoH verifies the classification of device and determines the cost of evaluation. The result of pre-registration along with the invoice is e-mailed to applicant. The local representative, on behalf of manufacturer shall make the payment and upload the proof of payment. MoH will review the documents and share the results through email to applicant. Some devices require In-country testing at an accredited Laboratory.
Regulatory Approval Process Overview
Freyr’s team of experts keep a track of changing trends and regulations and help the stakeholders in maintaining Regulatory compliance throughout the product lifecycle. We offer Regulatory solutions to maintain other Regulatory aspects of compliance within the constrained budgets.
Class of Device | Risk Class | MoH Timelines for Marketing Authorization | MoH Timelines for Renewal / Variation | ||
|---|---|---|---|---|---|
| Classification Process (Days) | Evaluation Process (Days) | Classification Process (Days) | Evaluation Process (Days) | ||
| Class A | Low Risk | 7 | 45 | 7 | 45 |
| Class B | Low Moderate Risk | 7 | 90 | 7 | 45 |
| Class C | Moderate – High Risk | 7 | 100 | 7 | 45 |
| Class D | High Risk | 7 | 120 | 7 | 45 |
Freyr Expertise
- Regulatory Due-Diligence
- Device Registration
- In-Country Testing
- Distributor Licensing
- Legalization and Notarization
- Legal Representative
- Labelling support
- Translation support
- Distributor identification and qualification
- Post Marketing Surveillance services
- Post Approval Change Management
- License renewal and transfer services
- Submission and liaising services
