
USFDA Medical Device Premarket Approval Overview
The USFDA Premarket Approval (PMA) process is one of the device registration pathways provided by the US FDA, primarily designed for FDA Class III medical devices. The FDA PMA approval process for Class III devices entails meticulous scientific and regulatory evaluations to assess the medical device's safety and efficacy, ensuring the highest standards are met prior to the market authorization.
Book a meeting with our premarket approval experts
Who Should Submit a USFDA Medical Device Premarket Approval (PMA) Application?
Device manufacturers must submit a PMA application if the device:
- Is novel.
- Belongs to a high-risk class.
- Cannot be found in the Product Classification Database.
- Is not substantially equivalent (NSE) to Class I, II, or III devices.
Get expert advice on your premarket approval Application
What is the Difference Between 510(k), PMA, and De-Novo Applications?
Premarket Approval
- Device pertaining to Class III that supports human life or that presents a potential, unreasonable risk of illness or injury.
- FDA PMA approval process requires clinical trials.
- Requires onsite inspection before issuing PMA approval.
- 180 calendar days
De-Novo Classification
- Novel devices of Class I and II that do not have a valid predicate device.
- Requires clinical study data.
- No onsite audit before De-Novo approval.
- 150 calendar days.
510(k) Registration
- FDA Class III devices that have substantial equivalency with the predicate device.
- It does not require human testing.
- No onsite audit before 510(k) clearance.
- 90 calendar days.
What are the Different FDA Premarket Approval Application Methods?
Manufacturers can opt for any of the following four (04) PMA application methods that would be best suited for their device:
- Traditional PMA
- Modular PMA
- Product Development Protocol
- Humanitarian Device Exemption
What are the Data Requirements for Medical Device Premarket Approval?
According to 21 CFR part 814, applicants must submit a duly filled CDRH application form, required undertakings, and a well-drafted PMA technical file to the US FDA. The technical file shall include the non-clinical and clinical data.
Non-clinical Data – It consists of data on microbiology, toxicology, immunology, biocompatibility, stress, wear, shelf life, and other laboratory or animal tests.
Clinical Data – It consists of data on study protocols, safety and effectiveness data, adverse reactions and complications, device failures, and replacements, patient information, patient complaints, tabulations of data from all individual subjects, results of statistical analyses, and any other information from the clinical investigations.
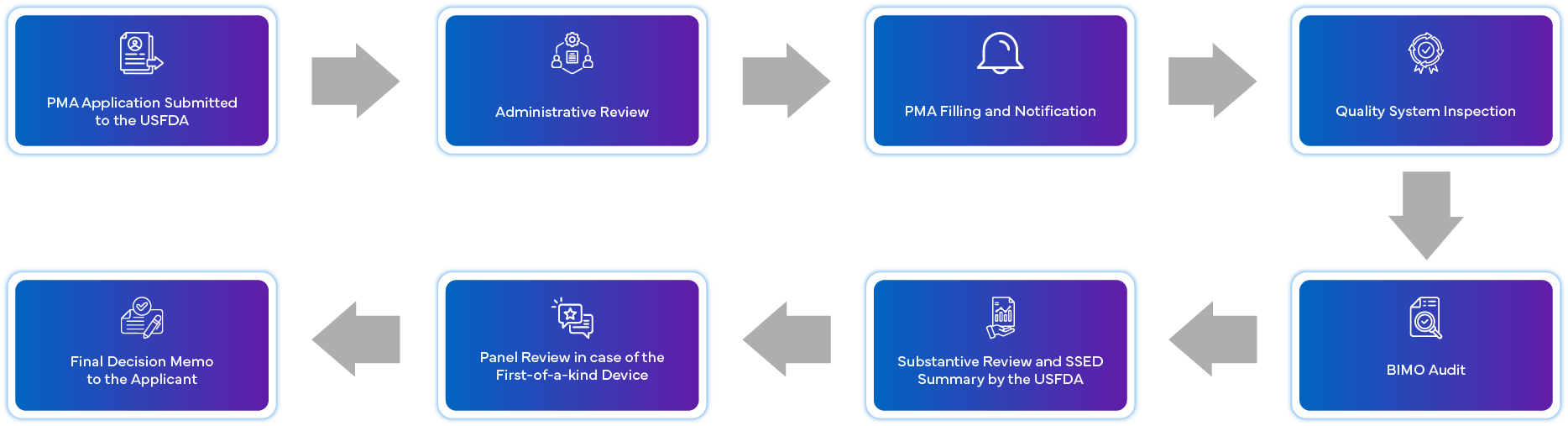
What is the PMA Application Process?
What are the Post Approval Compliance Requirements for PMA?
The devices approved under the PMA pathway shall comply with the post-marketing requirements set forth by the USFDA. The device must comply with the following:
- Post-approval requirements imposed in the FDA PMA approval order.
- Post-approval changemanagement through timely submission of relevant PMA supplements
- Submission for post-approval (annual) reports
- 21 CFR 803 regulations for Medical Device Reporting (MDR)
- Post-market Surveillance Studiesas required by the USFDA in the PMA approval orders.
What is the USFDA fees for reviewing the PMA application?
The MDUFA user fees for original PMA and supplements is as below-
| Application type | Fees for Fiscal Year 2023 (Oct 1st, 2022 through September 30th, 2023) | |
|---|---|---|
| Standard fee | Small business fee | |
| PMA, PDP, PMR, BLA | $441,547 |
|
| Panel-Track Supplement | $353,238 | $88,309 |
| 180-Day Supplement | $66,232 | $16,558 |
| Annual Fee for Periodic Reporting on a Class III device (PMAs, PDPs and PMRs) | $15,454 | $3,864 |
| 30-Day Notice | $7,065 | $3,532 |
| Real-Time Supplement | $30,908 | $7,727 |
With expertise in handling PMA submissions, Freyr can assist in identifying and compiling the information and assist in the preparation and review of the application.
USFDA Medical Device Premarket Approval Expertise and Advantages
- Regulatory Due Diligence
- Quality System Inspection Compliance
- BIMO Audit Compliance
- PMA Technical File Compilation
- Publishing and Creation of eCopy
- Validation and Submission of eCopy
- Addresses RTA Response and Deficiencies
- Liaising Services till FDA Premarket Approval
- Consultation for Deficiencies
- Device Listing and Establishment Registration
- Management of PMA Supplements and 30-day Notices
- Annual Periodic Reporting Submissions
- Mock Audits and 21 CFR 820 Training

- Experience in handling many FDA PMA submissions for diversified device categories.
- Expert team for FDA premarket approval application as per the regulatory requirements
- Additional support to handle PMA-related queries.
- On-time submission of deliverables
- Up to date with the US FDA new amendments
