
3 min read
Radiopharmaceuticals are pharmaceutical drugs that contain radioisotopes, which are used as diagnostic and therapeutic agents. They target specific organs, tissues, or cells in the human body. These medicinal products can be delivered orally, intravenously, or interstitially, and they form a subspecialty of radiation therapy. There are strict regulations for marketing radiopharmaceuticals, as they can be administered only by a professional specializing in nuclear medicine. However, the opportunities are immense due to their ability to diagnose certain medical problems or treat diseases such as cancer. The global radiopharmaceuticals market reached approximately US$5.9 billion by 2022, and it is expected to reach US$ 11.93 billion by 2030, growing at a Compound Annual Growth Rate (CAGR) of 11.76%.
Pharmaceutical companies from across the globe are trying to get a hold of their share of the radiopharmaceuticals market, including those in Canada. Health Canada (HC), the Canadian Health Authority (HA), is known for its efforts in making high-quality health services available to the public. Strict guidelines/regulations are in place to ensure patient safety first. Manufacturers/sponsors who wish to enter the Canadian radiopharmaceutical market must understand the regulations put forward by HC and accordingly design the ideal submissions strategy to obtain faster market approval.
The Regulatory Roadmap for Radiopharmaceutical Submissions in Canada
Any drugs marketed in Canada must follow the Food and Drugs Act (FDA) and the Food and Drugs Regulations (FDR). Radiopharmaceuticals are listed under Schedule C of the FDA. The Biologics and Genetic Therapies Directorate (BGTD), HC, regulates the market authorization of these products. They are approved for human use based on the satisfactory evaluation of the safety, efficacy, and quality of radiopharmaceutical submissions.
HC is an official member of the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH). It has adopted the ICH documents that relate to quality, safety, efficacy, and multidisciplinary topics. The list of these documents is available on the HC website for the sponsors’ reference.
In addition to the aforementioned documents, the Guidance for Industry: Management of Drug Submissions (guidance document), which was last updated on August 02, 2022, provides us with insights into HC’s perspective of evaluating the information submitted by sponsors on radiopharmaceuticals. The chart below (Figure 1) gives an overview of the topics covered in the guidance document.
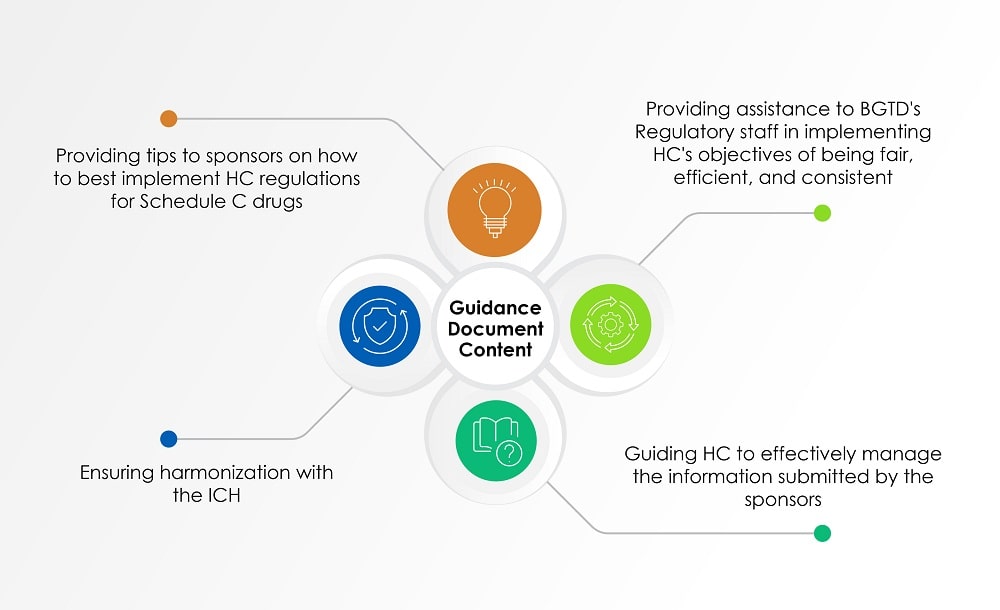
Figure 1: Contents of a Guidance Document
The Regulatory requirements for Schedule C drugs are the same as those for other drugs. The requirements include submissions and following the process for obtaining marketing authorization. However, the submission data requirements are different from other drugs because of their specific properties.
NDS
The drug manufacturer/sponsor must file a New Drug Submission (NDS) to the BGTD, which should contain the following data:
- Information on the drug’s safety, quality, and efficacy.
- Reports/results of pre-clinical and clinical trials.
- Information on the manufacture of the drug.
- Labeling and packaging particulars.
- Therapeutic claims and potential side effects of the drug.
The sponsor can make a presentation before the BGTD before submitting the NDS. Such a pre-submission meeting is beneficial, as the sponsor can address any gaps/queries raised by the BGTD and ensure that the submission contains all the required information. The meeting should ideally take place three (03) months before the due date of submissions.
Sponsors can also consider alternate review pathways such as a Priority Review or a Notice of Compliance with Conditions (NOC/c). Here is a summary of each of the two pathways:
- Priority Review: This pathway applies to an NDS or Supplement to a New Drug Submission (SNDS) for a life-threatening or severely incapacitating disease/condition for which there is enough evidence of the efficacy of the drug in terms of the diagnosis, treatment, or prevention of the said condition. It is also applicable when there is a substantial increase in efficacy and/or a considerable decrease in risk for a disease that does not have an effective drug in the Canadian market.
- NOC/c: This pathway considers the risk-benefit assessment of a drug. The NOC/c is for a drug that has an acceptable safety profile for a life-threatening or severely incapacitating disease/condition. Authorization is granted with favorable evidence for the radiopharmaceutical drug. This pathway applies to NDS, SNDS, and Abbreviated New Drug Submission (ANDS).
The NDS Format
HC has adopted the electronic Common Technical Document (eCTD) format for radiopharmaceutical drug submissions. In fact, separate Quality Information Summary templates such as QIS-R and QIS-PER have been created for radiopharmaceutical submissions.
Conclusion
While HC regulates the market authorization of radiopharmaceuticals based on the satisfactory evaluation of the safety, efficacy, and quality of the submissions, the Canadian Nuclear Safety Commission (CNSC) regulates the radiation safety of radiopharmaceutical drugs. CNSC controls aspects such as the handling, packaging, labeling, storage, disposal, etc., of radioactive materials. It also lays out regulations for the equipment used to manufacture radiopharmaceutical drugs.
Compliance with the current HC and CNSC regulations is essential for the satisfactory evaluation of radiopharmaceutical drug submissions. Partner with a proven Regulatory expert like Freyr for error-free and seamless Publishing and Submission activities in Canada. Stay informed! Stay compliant!









