3 min read
In the evolving world of medical devices, compliance isn’t a one-time task—it’s an ongoing commitment. Continuous monitoring and updates to key reports like Clinical Evaluation Reports (CERs), Performance Evaluation Reports (PERs), and Periodic Safety Update Reports (PSURs) are critical throughout the entire lifecycle of a medical device, from initial research to post-market surveillance. As the landscape of medical advancements and Regulatory requirements evolve continuously, ensuring the safety and compliance of medical devices and IVDs through effective medical writing continues to be the cornerstone of success and long-term viability.
Let’s look at how lifecycle management remains essential to a medical device’s success
The launch of a medical device is the culmination of years of effort spent on several phases like research, development, clinical trials, Regulatory submissions and post-market surveillance. This process spans over many years and each phase accumulates vital data in large quantity, that has to be carefully compiled and analysed to ensure that the device remains safe.
Compliance isn’t a one-time thing, and effective Lifecycle management helps avoid delays that may be costly later on. These delays often happen due to ineffective planning for unforeseen Regulatory changes or non-compliance, which may have been overlooked. Regular updates to critical documents like CERs, PERs, and PSURs ensure that manufacturers meet evolving Regulatory requirements and maintain product safety. Lifecycle management ensures that manufacturers, from the get-go, have planned for each stage, and thus they can avoid any pitfalls, and ensure successful market entry and longevity for their devices.
Are you keeping up with compliance?
From bandages to implants, the medical device industry is a cutting-edge, creative sector with a wealth of prospects. Despite the makers' strong desire to provide the market with safe and high-quality products, ambiguity nevertheless exists. The EU MDR replaced the Medical Device Directive (MDD) and the Active Implantable Medical Devices Directive (AIMDD). This regulation was implemented to impose more rigorous controls, improve patient safety, and promote increased transparency within the healthcare sector. There is a lot of uncertainty about those standards, and crucial compliance issues are frequently overlooked. However, medical device manufacturers should take steps to avoid Regulatory difficulties.
Let’s investigate the most common challenges faced by medical device manufacturers
- Maintaining CAPA (Corrective and Preventive Action)
- Adhering to Complaint Procedures
- Following Vigilance Procedures
- Clinical Evaluation and Post Market Surveillance
- Connecting with notified bodies
Best practices to remain compliant
To overcome these challenges, we at Freyr have established
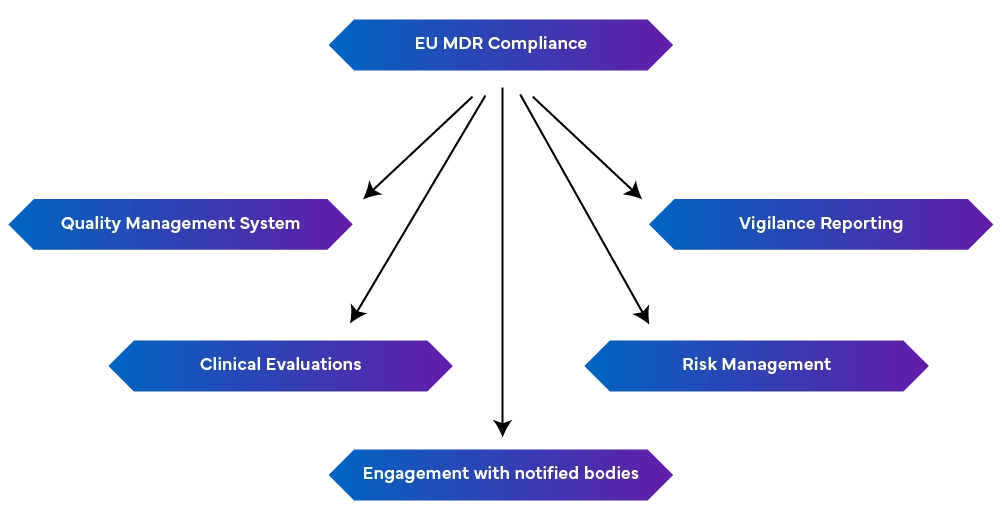
-a Robust Quality Management System (EN ISO 13485:2016): This includes production aspects, including Regulatory compliance, technical documentation, EU declarations of conformity, and risk management.
-Vigilance Reporting: Maintaining vigilance reporting as an ongoing process rather than a one-instance effort.
- Proactive Risk Management: This identifies and mitigates risks throughout the device lifecycle by regularly updating to address emerging risks.
- Conduct Clinical Evaluations and Post-Market Surveillance: This is done by demonstrating device safety and performance using clinical data. If required, clinical investigations should be performed, and clinical evaluations should be regularly updated with post-market surveillance data. Periodic safety update reports summarizing findings are also required.
- Connected with Notified Bodies: Changes in regulations can affect MDR requirements, making it crucial to maintain a good relationship with Notified Bodies for prompt updates and modifications.
How a Regulatory expert can help you
Trying to keep up with an ever-changing Regulatory landscape can be tough. So why not let an expert guide you through this maze? Managing the Regulatory demands of a medical device’s lifecycle is not only complex but resource intensive. In case of smaller companies, where internal resources may be better utilized in other areas, the help of an external Regulatory partner may prove indispensable. They offer specialized knowledge of evolving regulations and ensure that your key reports—such as CERs, PERs, and PSURs—are continuously updated and in compliance.
You can avoid costly delays, prevent non-compliance, and streamline your approval process. Additionally, a Regulatory expert can offer tailored solutions based on your device’s specific needs, ensuring that post-market surveillance, gap analysis, and all compliance documentation are up to date. Their involvement can make the overall process simpler, minimize the risks, and ensure that the device can meet the safety and performance expectations.
Freyr can help you with all your Regulatory needs concerning lifecycle management. Reach out to us today!