
5 min read
Submissions scheduled for Sep 24, 2016.
Now that the second phase of UDI compliance, for Class III I/LS/LS medical devices, has been implemented, many device manufacturers, especially of Class II type, are questioning how best they could be prepared for September 24, 2016 Class II device data submission deadline. To give them heads up, at Freyr, we have identified some of the prerequisites they should consider to line up Class II devices for compliance with FDA’s UDI mandate.
The new regulation requires all class II medical devices to be labeled and packaged with a unique device identifier (UDI) and entered into the FDA’s Global Unique Device Identification Database (GUDID). Given the volatility of compliance requirements coupled with shorter submission timeframes, the challenge for device manufacturers is to get to know the nitty-gritty of the compliance processes. At the same time, they must ensure that none of the key device attributes is missed-out while pulling together the scattered device data across different systems and reconciling it in spreadsheets to create compliance reports.
In order to assist device manufacturers easily navigate through this time-critical and complex compliance process, with no errors, Freyr has compiled the following pre-requisites to be followed.
Determine the UDI Compliance Date: Since the time FDA has issued its final rule, some of the device compliance dates have been changed and extended. To meticulously plan ahead the compliance strategies and processes and to avoid hasty last-minute amendments, labelers shall determine the accurate compliance date.
Class II Devices Compliance Date Compliance Requirements Sep 24 2016Class III devices required to be labeled with a UDI must bear a UDI as a permanent marking on the device itself if the device is a device intended to be used more than once and intended to be reprocessed before each use. The labels and packages of class II medical devices must bear a UDI Dates on the labels of these devices must be formatted as required Class II stand-alone software must provide its UDI as required Data for class II devices that are required to be labeled with a UDI must be submitted to the GUDID database For most devices, the compliance date for direct making is different than the other requirements. Basing on your product category, either intended to be reused or intended to be reprocessed, determine the Direct marking UDI compliance date as shown here:
Direct Marking Compliance Dates Category of Device – Reused and Reprocessed Sep 24 2015 Life-sustaining and life-supporting devices, regardless of device class Sep 24 2016 Class III devices and devices licensed under the Public Health Service Act Sep 24 2018 Class II devices Sep 24 2020 Class I devices and unclassified devices Evaluate the need of Direct Marking of the UDI number: All medical devices which are used more than once or which are to be reprocessed before each use must have direct marking of the UDI. The exception is for implantable devices which do not require direct marking as per the UDI rule. The devices which are for single-use, even if re-processed, are also not required to bear a permanent UDI – 21 CFR 801.45(d)(3). Thus, evaluate the need of Direct Marking basing on the category of medical devices you manufacture.
- Plan for Comprehensive Compliance: Review the FDA requirements for your specific products to be complied. Perform a through gap analysis to find out shortfalls related to data or technology to deal with some of the major challenges in the process of meeting the strict FDA timelines. Some of the challenges could be obtaining the DI or PI information, and handling large volumes of unstructured data from disparate sources, etc. Instead of burning midnight oil to reconcile all medical device data at the nick of the time, plan out for comprehensive compliance ahead of time through validated systems and tools which support data integration, data quality and data management.
![]()
Obtain DI number and Agency Membership: The UDI is composed of Device Identifier (DI – unique number based on version or model of device) and Product Identifier (PI – includes lot number, serial number, or expiration date). The DI portion of UDI will serve as the primary key to look up information about the device in the GUDID. To assign the DI, the FDA has accredited three Issuing Agencies – GS1, HIBCC, and ICCBBA. In this scenario, labelers must obtain membership of one of the agencies to get the DI number which needs to be entered into the FDA’s GUDID.
![]()
Identifier Attributes Issuing Agencies UDI DI (Device Identifier – Static Data)
Required to be synched to GUDIDUnique Number of
Manufacturer
Device Make
Device Model
GSI
HIBCC
ICCBBAPI (Product Identifier – Dynamic Data)
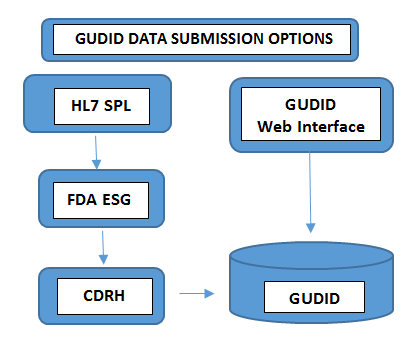
Required on all levels of packagingLot Number, Serial Number, Manufacture Date Expiration Date, - Submit the Data: The way you submit the data to GUDID varies with the volume of product portfolios you handle. Device manufacturers with minimum number of devices choose to submit the UDI information manually through FDA’s free GUDID web interface. In this case, only one DI record could be submitted at a time through a secured GUDID web interface.In the other case, manufacturers with larger number of product portfolios choose HL7 SPL submission option to collect data electronically and convert the consolidated data to SPL format before submitting it to the FDA’s Electronic Submission Gateway (ESG), using the DUNS number.Kindly make a note that GUDID account is not by submission type. The account is to identify you, the labeler, to enable submission of device data through both the options.
![]()
- Set up a GUDID Account: A labeler / device manufacturer requires one or more GUDID accounts based on the number of roles to be allocated; to name a few, GUDID coordinator, Data Entry User etc. But to authorize each role for data entry, manufacturer needs an approval from the FDA prior to account creation. The process of creating an appropriate GUDID account involves sending an email request to FDA upon which the applicant, you, will receive an account request document to fill out. Once you send back the filled up document to the FDA via email, the agency will review the form, and then sends an email with GUDID account log in information.


UDI implementation is a complex and time consuming process. During the course, while meeting with FDA’s UDI requirements, medical device manufacturers face many challenges pertaining to data management, data integration and data submission. With compliance deadline for Class II devices just a year away, Freyr recommends that companies start working towards it right now.
To navigate your organization through this complex compliance process, Freyr offers the best of both worlds – on-demand, fully configurable, UDI software solution, Freyr IDENTITY, as well as a Centre of Excellence (CoE) that offers best in class, cost-effective and customizable UDI services built around your unique and demanding requirements.









