
2 min read
After nearly two (02) decades of issuing the first draft guidance, the United States Food and Drug Administration (US FDA) finalized the guidance on population Pharmacokinetics (PK) in February 2022. The latest draft explains the application of population PK in drug development and recommendations on therapeutic individualization. The guidance applies to the Investigational New Drug (IND) applications, New Drug Applications (NDAs), Biologics License Applications (BLAs), and the Abbreviated New Drug Applications (ANDAs).
The data from population PK analysis must be included by sponsors/drug manufacturers in marketing applications to simplify the post-marketing activities. They can also be crucial in answering Regulatory questions from the Health Authorities (HAs).
Understanding Population PK
The study of the variability in drug concentrations within a patient population receiving clinically relevant doses of a specific drug is defined as population PK. It aims to identify and quantify the variability sources that are useful in the manufacturing and recommending doses of the said drugs.
The following illustration can be referred to for understanding the whole process of population PK:
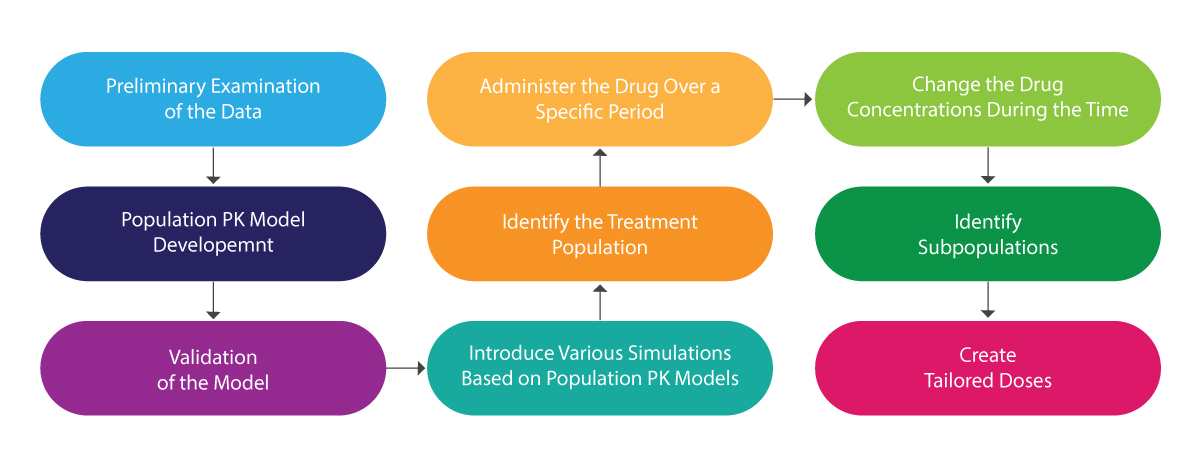
Population PK Analysis – How it Helps?
- Helps in designing customized dosage regimes.
- Provides a reliable estimation of the covariate (a variable whose change can be predicted in a research/study and can be used to predict the outcome of the said study).
- Considers factors such as demography, environmental, biological, concomitant medicines (other drugs that the patients are taking during the trial), and the drug concentration.
- Useful in designing safer and qualitative clinical trials, including pediatric.
- Obtain exposure matrix for performing exposure-response analysis.
An Overview of the US FDA’s Final Guidance on Population PK
The final draft gives an insight into the FDA’s current thinking on the data from population PK analysis and model submissions to help make Regulatory decisions. Here is a brief overview of the draft:
Labeling - It provides recommendations on incorporating the apt information from the said analysis in labeling. The results have to be presented in the ‘Clinical Pharmacology” section and are to be summarized in other sections of labeling, as required.
Content - The content and relevant format to be followed for the submission of population PK reports to the FDA have also been detailed.
Regulatory decision-making – The population PK reports that are useful to make Regulatory decisions have to be included in Module 5 of the electronic Common Technical Document (eCTD).
Going Forward
Sponsors who need advice on the use of population PK analysis in their drug development process or in answering Regulatory questions have been asked to do so at their important meetings with the Agency. Furthermore, it is imperative that good model performance is ensured for accurate results. This can be accomplished by using sufficient PK data on the specific patient population and relevant subpopulations.
It can be challenging for drug manufacturers/sponsors to submit accurate population PK reports in the eCTD format and follow labeling regulations for compliance. Reach out to a Regulatory expert like Freyr, who can assist you with your submissions and Regulatory labeling activities.









