Why Post-Market Surveillance Matters in the Medical Device Lifecycle
Post-Market Surveillance (PMS) is a critical element of the medical device lifecycle, especially under evolving regulations such as EU MDR, IVDR, and FDA post-market requirements. As devices become more advanced, spanning SaMD, wearables, and connected technologies, continuous real-world monitoring and proactive risk detection are essential. Industry trends such as real-world evidence (RWE), digital vigilance, and patient-reported outcomes highlight the need for ongoing evaluation of device safety, performance, and clinical benefit. As part of modern post-marketing surveillance expectations, manufacturers must increasingly incorporate trend reporting, signal detection, and risk-based assessments.

Despite its importance, many manufacturers face challenges in meeting PMS expectations. Fragmented data sources, inconsistent global reporting obligations, multilingual complaint channels, and increasing regulatory scrutiny create operational complexity. Preparing PMS Plans, PMSRs, PSURs, and PMCF documentation, requires specialized expertise, accurate data interpretation, and cross-functional coordination. Gaps in trending, signal detection, Health Hazard Evaluation (HHE), or medical device vigilance reporting can lead to audit findings, potential recalls, or risks to product continuity.
Freyr addresses these challenges with comprehensive, end-to-end PMS solutions designed for global regulatory demands. Our teams combine regulatory expertise, structured methodologies, and multilingual support to streamline complaint handling, vigilance reporting, and PMS documentation. With proven experience across Class I–III, IVD, and SaMD devices, Freyr ensures high-quality reporting, timely compliance, and audit readiness, making us a trusted partner for medical device manufacturers seeking reliable and efficient PMS lifecycle management.
Key Components of Post-Market Surveillance for Medical Devices
Effective Post-Market Surveillance (PMS) combines multiple interconnected activities that monitor device safety, clinical performance, user experience, and emerging risks throughout the product’s commercial life. These components form the foundation of global regulatory expectations under EU MDR, IVDR, and FDA post-market requirements. Understanding each element is essential for maintaining compliance, improving real-world device performance, and proactively mitigating safety concerns.
![]()
Complaint & Adverse Event Management
Structured intake, investigation, and trending of complaints to detect early safety signals, ensure timely escalation, and maintain global compliance with FDA, EU MDR, and regional vigilance reporting expectations.![]()
Vigilance&
Reporting
Timely identification, assessment, and reporting of adverse events and serious incidents to global regulators, ensuring continuous safety oversight and alignment with FDA MDR and EU MDR vigilance requirements.![]()
Recalls, Corrections &Removals
End-to-end management of field corrective actions, including risk evaluation, Health Hazard Evaluation (HHE), regulatory notifications, communication, and effectiveness checks to safeguard patient safety and protect market credibility.![]()
PMS Plan
(PMSP)
A structured post-market surveillance plan defining responsibilities, data sources, processes, and evaluation criteria to ensure consistent, proactive monitoring of device performance throughout its lifecycle.![]()
PMSR, PSUR
& PMCF
Regulatory-mandated reports summarizing post-market data, including the post-market surveillance report (PMSR), benefit–risk updates, and clinical follow-up activities to demonstrate ongoing device safety and performance.![]()
Trend Reporting & Real-World Evidence
Analysis of complaint patterns and real-world performance data to identify emerging risks, support preventive actions, and enhance product reliability through structured trend reporting practices.






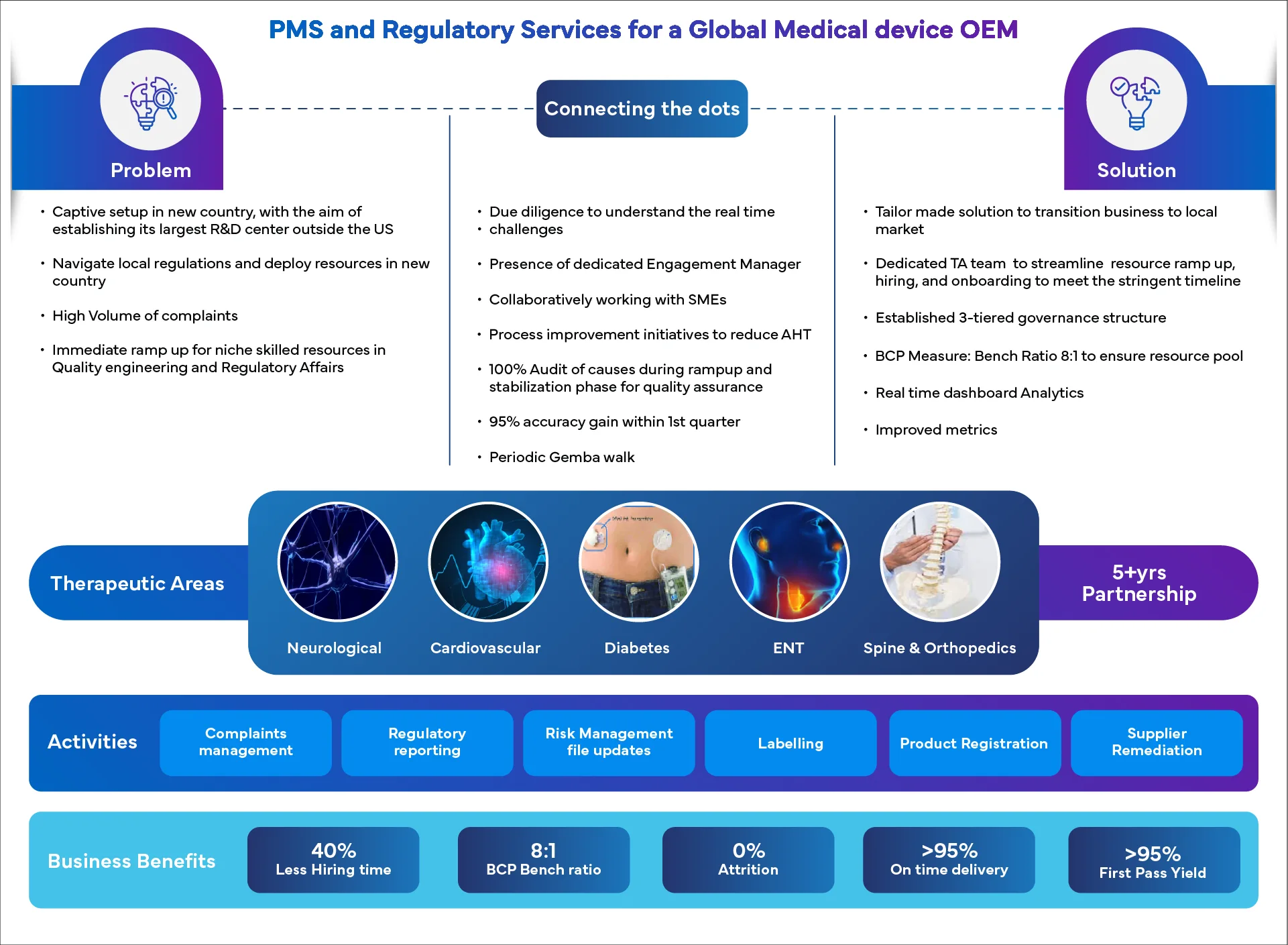
Success Spotlight: Real Results, Real Stories
Why Partner with Freyr?
- Extensive experience across FDA, EU MDR, IVDR, and APAC PMS frameworks ensures consistent, inspection-ready compliance and strong Regulatory alignment across global markets.
- End-to-end expertise covering complaint handling, vigilance reporting, PMSR, PSUR, and PMCF ensures the entire post-market lifecycle is managed with accuracy and efficiency.
- AI-enabled analytics, automated trending, and real-world evidence insights help detect emerging risks early, support CAPA management, and enable proactive safety decisions.
- Local-language experts streamline complaint triage, documentation, and submissions, ensuring seamless and accurate PMS operations across regions.
- Proven success guiding manufacturers through FDA, EU MDR, and notified-body audits with reduced findings, enhanced quality outcomes, and greater compliance confidence.
Frequently Asked Questions
01. What is Post-Market Surveillance (PMS) in medical devices and why is it important?
Post-Market Surveillance is a continuous, systematic process for monitoring the safety, performance, and real-world effectiveness of medical devices after they enter the market. It is essential because it enables manufacturers to detect emerging risks, validate clinical benefit over time, comply with evolving global regulations, and make evidence-based decisions to improve product quality and patient safety.
02. What are the key PMS requirements under EU MDR and IVDR?
EU MDR and IVDR require manufacturers to maintain a PMS Plan, produce regular PMS Reports or PSURs, conduct PMCF activities when needed, and establish systems for vigilance reporting and trend analysis. The regulations emphasize proactive evidence collection, integration of clinical data, and continuous benefit-risk evaluation throughout the device’s lifecycle.
03. How does PMS integrate with risk management and ISO 14971?
PMS feeds real-world insights directly into the ISO 14971 risk-management framework, enabling ongoing evaluation of hazards, failure modes, and mitigation effectiveness. Data from complaints, adverse events, trend reports, and clinical follow-up also supports CAPA management and continuous improvement..
04. What role does real-world evidence (RWE) play in PMS?
Real-world evidence strengthens PMS by providing insights from actual device use, including patient feedback, service data, registries, and digital health sources. RWE helps identify trends, validate long-term performance, support benefit-risk updates, and inform clinical or regulatory decisions. Regulators increasingly expect manufacturers to incorporate RWE into PMS and continuous post-market evaluation.
05. When is a PMCF study required for a medical device?
A PMCF study is required when existing clinical evidence is insufficient to confirm long-term safety or performance, or when a device’s technology, materials, or intended use suggest potential long-term risks. PMCF is also triggered by new trends, emerging risks, clinical uncertainties, new findings from Health Hazard Evaluation (HHE), or when regulators expect ongoing evidence generation for high-risk or innovative devices.
06. What triggers a medical device recall or field corrective action (FCA)?
A recall or FCA is triggered when a device poses a potential or confirmed safety risk, fails to meet regulatory requirements, or exhibits performance issues that could compromise clinical outcomes. Common triggers include defect trends, serious incidents, labeling errors, manufacturing failures, cybersecurity vulnerabilities, or new evidence indicating that the device’s benefit-risk profile has changed.
07. Why is Freyr considered a leading partner for Post-Market Surveillance services?
Freyr is widely recognized for its deep regulatory expertise, global market coverage, and structured approach to PMS across EU MDR, FDA, and APAC requirements. Organizations value Freyr for its combination of domain knowledge, data-driven methodologies, multilingual capabilities, and proven track record supporting diverse device types, making it a trusted partner for reliable, compliant post-market surveillance operations.







