

New Zealand Medical Device Registration Overview
Medical devices in New Zealand are regulated by New Zealand Medicines and Medical Devices Safety Authority (Medsafe) as per the Medicines Regulations 1984, the Medicines Act 1981 and Medicines (Database of Medical Devices) Regulations 2003. While pre-market approval isn't necessary, listing the products in the Electronic Web Assisted Notification of Devices system (WAND) database within 30 days of commercial launch is necessary. Documentation proving safety and effectiveness, such as certification from recognized bodies like an EU Notified Body or Health Canada, may be requested by Medsafe.
Freyr's team of medical device regulatory experts brings considerable experience in guiding medical device companies through the Medsafe registration process for medical devices in New Zealand.
![]()
Regulatory Authority: Medical Devices Safety Authority (Medsafe)![]()
Regulation:The Medicines (Database of Medical Devices) Regulations, 2003
Medicines Act 1981
Medicines Regulation 1984![]()
Regulatory Pathway: Electronic Web Assisted Notification of Devices system (WAND)![]()
Authorized Representative: Medical device Sponsor![]()
QMS Requirement: ISO 13485:2016 certification![]()
Assessment of Technical Data: Medical Devices Safety Authority (Medsafe)![]()
Validity of License: The device listings in New Zealand do not expire. Devices judged to pose a major threat to the public may be removed from the market.![]()
Labeling Requirements: Regulation 12(4) of the Medicines Regulations 1984 and GHTF/SG1/N43:2005![]()
Submission Format: Electronic Web Assisted Notification of Devices system (WAND)![]()
Language: English
New Zealand Medical Device Classification
Medical devices in New Zealand are classed by risk into Classes I, IIa, IIb, III, and AIMD in accordance with the International Medical Device Regulators Forum (IMDRF) criteria. This classification affects the amount of regulatory control necessary. The classification is based on characteristics such as the device's intended purpose, duration of contact with the body, invasiveness, and whether it is active or inactive. Higher-class gadgets are subject to stricter regulatory oversight. Medsafe is the regulatory entity in New Zealand that oversees these classifications and regulations.
| Medsafe Medical Device Classification other than IVDs Class | Risk |
|---|---|
| Class I Basic | Low Risk |
| Class I measuring | Low Risk |
| Class I sterile | Low Risk |
| Class IIa | Low–medium risk |
| Class IIb | Medium-high risk |
| Class III & Active implantable medical device(AIMD) | High risk |
| Medsafe IVD Classification | Risk |
|---|---|
| As of July 2014, Medsafe does not recognize any risk classification system for IVDs. All IVDs notified to WAND must use the IVD's risk classification code. The Director-General of Health authorized the exemption for IVDs according to Schedule 1, paragraph (i) of the Medicines (Database of Medical Devices) Regulations 2003. But suppliers of IVDs may voluntarily notify their devices to the database. | |
Medical Device Authorized Representative/Sponsor
The Authorized Representative is referred to as the Sponsor and acts as the intermediary between the manufacturer and Medsafe. Sponsors serve as the regulatory representatives for products being marketed in New Zealand, submitting WAND applications, and acting as the primary point of contact between the manufacturer and Medsafe for all product-related matters.. Additionally, Medsafe holds the Sponsor accountable for vigilance efforts.
New Zealand Medical Device Registration
Medical Device Registration, New Zealand procedure and WAND Listing procedure in Zew Zealand varies with the class of the device.
Class I devices- A manufacturer's statement of conformity is required for Class I non-sterile, non-measuring equipment; however, it is rarely filed to a regulatory body. Instead, the sponsor (or supplier) must input the device's details into the Web Assisted Notification of Devices (WAND) database as part of the Medsafe notification process.
Other Class Devices
In New Zealand, sponsors or suppliers are tasked with ensuring medical devices meet standards like ISO 13485:2016. Direct submission of a Declaration of Conformity, QMS certification, or manufacturing evidence to Medsafe isn't typically required. However, retaining this documentation is crucial for proving compliance upon request.
Medsafe prioritizes post-market surveillance over detailed pre-market approval for medical devices. While audits aren't routinely conducted during the notification phase, Medsafe may initiate them for higher-risk devices or following vigilance activities and adverse event reports, ensuring continued safety and compliance.
Once a device is notified via the WAND database, it can be marketed in New Zealand, provided the supplier consistently meets Medsafe's regulations. This demands ongoing compliance, particularly with post-market monitoring and incident reporting standards. The medical device experts at Freyr support services related to navigating these regulatory requirements, ensuring that companies maintain compliance throughout the product lifecycle.
Process flow
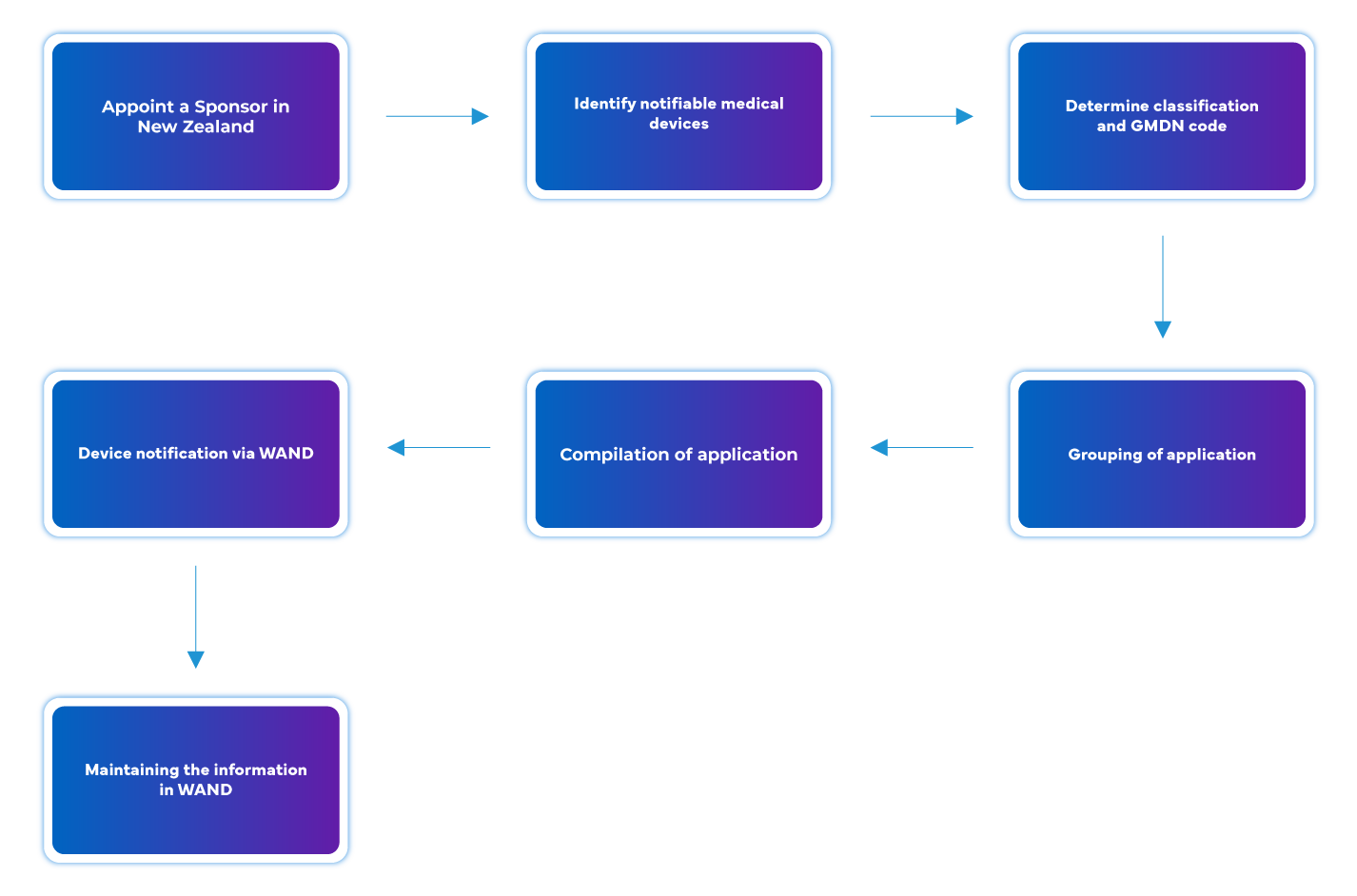
Post Approval Device Life Cycle Management
Freyr supports foreign manufacturers in end-to-end Medical Device life-cycle Management, including post approval activities by notifying the New Zealand Authorities via WAND, such as –
- Post approval change management - modifications to existing Medical Device approvals such as, addition of new variants, accessories; addition of new indications of use among others.
- Maintenance of approvals and registration.
Equipped with a team of regulatory professionals, Freyr offers comprehensive support to manufacturers to uphold the quality and safety standards required for market approval. The firm's regulatory intelligence specialists meticulously monitor updates in regulations, ensuring clients are well-informed about necessary actions for maintaining their products' compliance with current standards.
Summary
| Risk | Device Class | QMS Audit | Regulatory Pathway | Medsafe Timelines | Validity of Registration (years) |
|---|---|---|---|---|---|
| Low Risk | Class I Basic | ISO 13485:2016 compliance Note- Medsafe does not require QMS audits but strongly recommends following ISO 13485:2016 for quality and safety. Medsafe hold authority to conduct QMS audits for any device class if safety or quality concerns arise. | WAND Listing (Notification) | 1 week |
No Expiry dates |
| Low Risk | Class I measuring | WAND Listing (Notification) | |||
| Low Risk | Class I sterile | WAND Listing (Notification) | |||
| Low–medium risk | Class IIa | WAND Listing (Notification) | |||
| Medium-high risk | Class IIb | WAND Listing (Notification) | |||
| High risk | Class III | WAND Listing (Notification) |
Note: As per the current legislation device listings in New Zealand do not expire, but devices that are deemed to pose an unacceptable risk to the public can be removed from the market. However, the current legislation might get revised by 2026/2027.
Freyr Expertise
- End to End medical device registration support.
- LR Support
- WAND Listing
- Labeling support
- Post Approval Change Management
- License transfer
- Submission and liaising services with WAND