
4 min read
With the US Food and drug administration (USFDA) implementing the final rule for the Quality management system regulations (QMSR) in 2024, medical device manufacturers must embrace the amendments to market and distribute their devices in the USA market.
This rule updates the USFDA’s Quality System Regulations (QSR) by aligning with the ISO 13485:2016, which is the international standard for medical device QMS. Medical device manufacturers have two-year transition period to conform, which makes it necessary for the organizations to adapt the new requirements to avoid noncompliance at the time of inspection.
What is QMSR?
The FDA QMSR is a streamlined approach to QMS requirements, which is an update to the former QSR structure. This alignment is crucial because it simplifies global compliance for the manufacturers specially those who are operating globally. This harmonization will allow companies to meet Regulatory requirements both in the US and other markets in a much more unified manner.
QMSR mandates improvements in, Risk management, device design, post market surveillance. This role may add more complexity, but it also provides the manufacturers to standardize their quality procedures, which in turn increases the device safety, also increase better documentation, which can be vital during USFDA inspections.
Key changes in the QMSR
- Harmonization with ISO 13485: This is the most critical step which allows medical device manufacturers to accept internationally accepted standards. The USFDA recognized that many medical devices company already conform with ISO 13485, which reduces duplicative efforts.
- Risk Management’s emphasizes risk management throughout the lifecycle of the medical device. Medical device manufacturers must exhibit effective risk management, risk control and risk management.
- Device design and controls: Under QMSR, design controls extended to ensure that medical device manufacturers fully account for user needs, safety of the device and performance criteria, which is a focus area during USFDA inspection.
- Post Market Surveillance: Companies need to enhance the post market monitoring system. This will expect manufacturers to gather information on the device safety and effectiveness which help in finding the issues quickly and identified quickly.
- Documentation and record keeping: This is the final rule which stresses on the documentation. Thorough / proper recordkeeping is critical during the inspection.
Steps to prepare for US Food and Drug Administration inspection:
With the inspection lasting until 2026, companies have two years for their quality systems which aligns with QMSR. However, waiting for the last minute can be risky.
Steps for USFDA inspection under QMSR framework for industries where current QMS is based out of QSR:
- Conduct a Gap analysis: This is the first step where the current quality system diverges from new QMSR requirements. A through gap analysis will help identify the areas which need updates such as risk management, post market surveillance and design controls.
- Update risk management procedure: Ensure that risk management activities are combined with the entire lifecycle of your product from design to post market surveillance.
- Revisit design control: Manufacturers should ensure that the design process is robust and well documented. Validate the design process are robust and well documented and fully integrated into your quality management system.
- Enhance Post market surveillance: Implement systems to monitor the performance of the device after they are placed in the market. It could involve setting customer feedback mechanisms, gathering clinical data evidence and tracking them closely.
- Training and documentation: Training the staffs on the new requirements, especially those who are involved in the quality management and Regulatory compliance. This ensure that all documentation process is aligned with QMSR expectations.
- Third party certification: If your company is not ISO 13485 certified, now might be the time to consider. Achieving ISO 13485 certification can give you a head start in meeting the requirements by USFDA to boost credibility in global markets.
Navigating the Two-year transition period
To effectively utilize the transition period and ensure compliance with the USFDA's Quality Management System Regulation (QMSR), manufacturers should adopt a proactive approach. Here's a suggested roadmap:
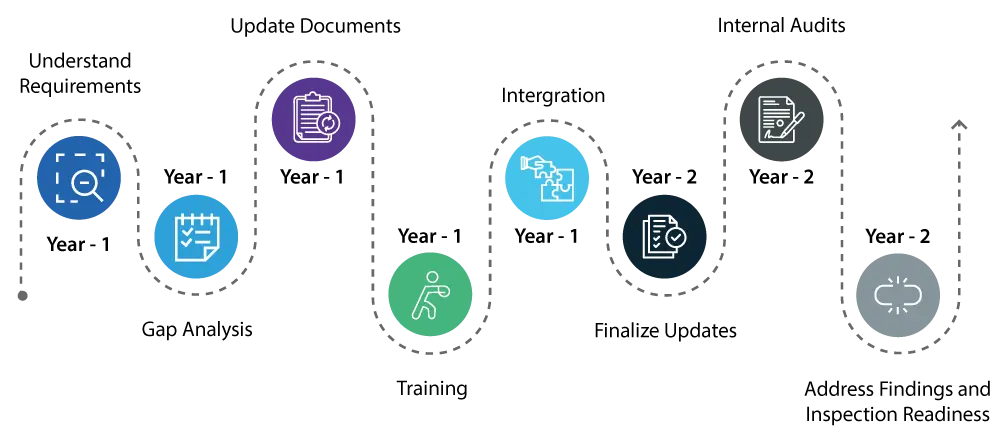
Year 1:
- Understand the requirement: Start by thoroughly understanding the changes introduced by the QMSR. This involves a detailed review of the new requirements and how they differ from the existing Quality System (QS) Regulation.
- Know the gaps : Conduct a comprehensive gap analysis to identify areas within your current quality system that require updates to meet the new QMSR standards.
- Document updating / remediation : Initiate the necessary updates to your quality system, focusing on areas such as risk management and design controls, which are critical components of the QMSR.
- Training :Begin training your staff on the new regulations to ensure that everyone involved is aware of the changes and understands their roles in maintaining compliance.
- Integration : Start integrating the new QMSR requirements into your daily operations to make the transition smoother.
Year 2:
- Continue with the implementation of changes to your quality system, ensuring that all updates are fully integrated and operational.
- Conduct thorough internal audits to verify that the updates are effective and that your quality system is fully aligned with the QMSR requirements.
- Address any findings from the internal audits promptly to ensure that all aspects of your quality system are compliant.
- By the end of the second year, your quality system should be fully compliant with the QMSR, and you should be prepared for USFDA inspections with confidence that there will be no major issues.
By following this roadmap, manufacturers can not only meet the USFDA's requirements but also establish a robust quality system that is efficient, standardized, and recognized globally. This proactive approach will help ensure a smooth transition to the new regulations and maintain the highest standards of quality and safety for medical devices.
Conclusion: Taking Proactive Steps
Preparing for USFDA inspections under the new QMSR rule is not just about avoiding penalties—it’s about improving the device safety and effectiveness. By harmonizing with ISO 13485, the USFDA is setting higher expectations, but also providing a pathway to more streamlined global compliance. Manufacturers who start to adapt early, should focus on key areas like risk management and post-market surveillance, and to ensure their quality systems are up to date and robust, that will not only meet Regulatory expectations but also enhance their competitive edge in the market.
